Next: CONCLUDING REMARKS Up: STOCHASTIC MOLECULAR OPTIMIZATION USING Previous: THOR - MOLECULAR FORCE
![]()
![]()
![]()
Next: CONCLUDING
REMARKS Up: STOCHASTIC
MOLECULAR OPTIMIZATION USING Previous: THOR
- MOLECULAR FORCE
We have applied the SMO to investigate conformations of
some molecular systems. The first application of our approach is to search
the range of ![]() that answer the question ''Which
that answer the question ''Which ![]() or distribution of probability correspond to a minimum number of steps
to find the global minimum energy ?''.
or distribution of probability correspond to a minimum number of steps
to find the global minimum energy ?''.
To answer this question we have generate two maps (Figs.
2-3) in terms of ![]() nC is the number of cycles to obtain the convergence in the energy. That
is, for each (q,T) we calculate the number of steps necessary to search
the global minimum in case of H
nC is the number of cycles to obtain the convergence in the energy. That
is, for each (q,T) we calculate the number of steps necessary to search
the global minimum in case of H ![]() O and H
O and H ![]() O
O ![]() molecules.
molecules.
H2O (Dimension D=1)
Using fixed H-O distance we scan the angle ![]() (D=1) between H-O-H bonds to investigate the angular energy (rotational
barrier) for the H2O molecules. If we scan the potential surface in 0.1
(D=1) between H-O-H bonds to investigate the angular energy (rotational
barrier) for the H2O molecules. If we scan the potential surface in 0.1 ![]() steps this results in 3600 cycles necessary to search the angular range
(0
steps this results in 3600 cycles necessary to search the angular range
(0 ![]() 360
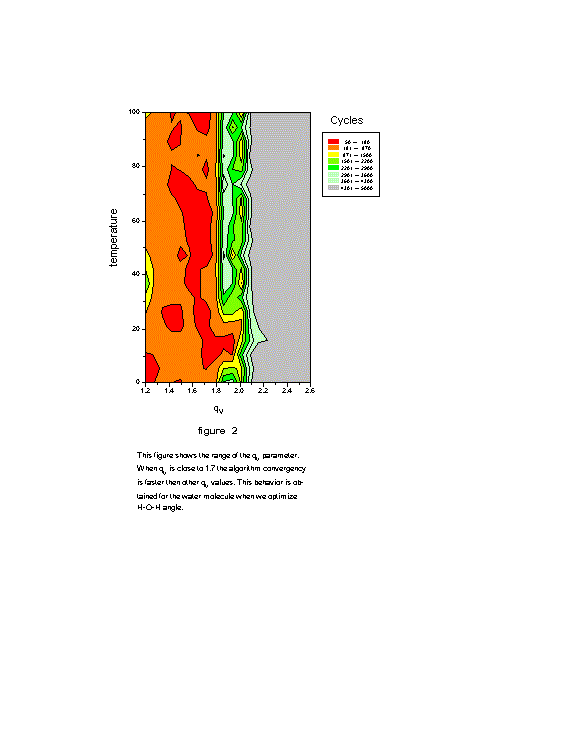
360 ![]() ). However, with the random procedure proposed by GSA algorithm we obtain
the global minimum with less 180 cycles for (q
). However, with the random procedure proposed by GSA algorithm we obtain
the global minimum with less 180 cycles for (q ![]() ; q
; q ![]() )=(1.5; 1.7) machine (Fig. 2). When q
)=(1.5; 1.7) machine (Fig. 2). When q ![]() is close to 1.7 the algorithm convergence is faster than other q
is close to 1.7 the algorithm convergence is faster than other q ![]() values.
values.
H2O3 (Dimension D=2)
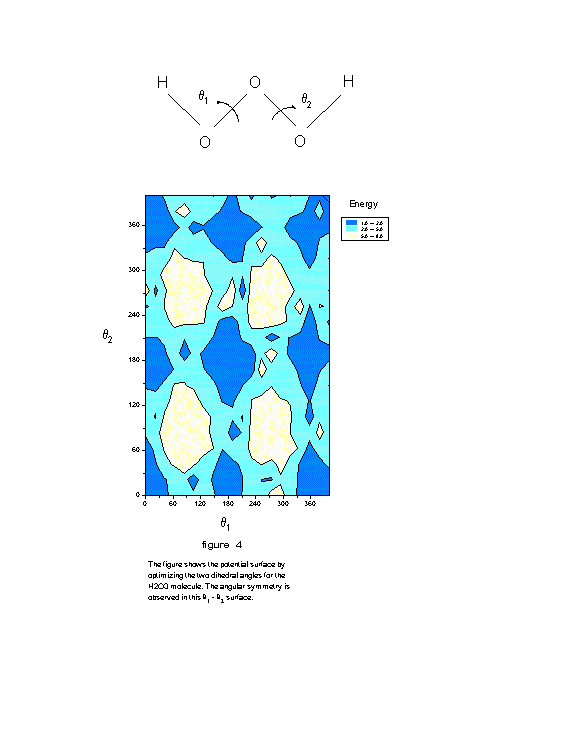
For the H ![]() O
O ![]() molecule we have optimized two (D=2) proper dihedral angles
molecule we have optimized two (D=2) proper dihedral angles ![]() and
and ![]() (Fig. 3 - 4) and obtained
the global minimum with less then 840 cycles using (q
(Fig. 3 - 4) and obtained
the global minimum with less then 840 cycles using (q ![]() ; q
; q ![]() )=(1.5; 1.6) machine (we point out that 3600x3600 cycles is far greater
than 840 cycles).
)=(1.5; 1.6) machine (we point out that 3600x3600 cycles is far greater
than 840 cycles).
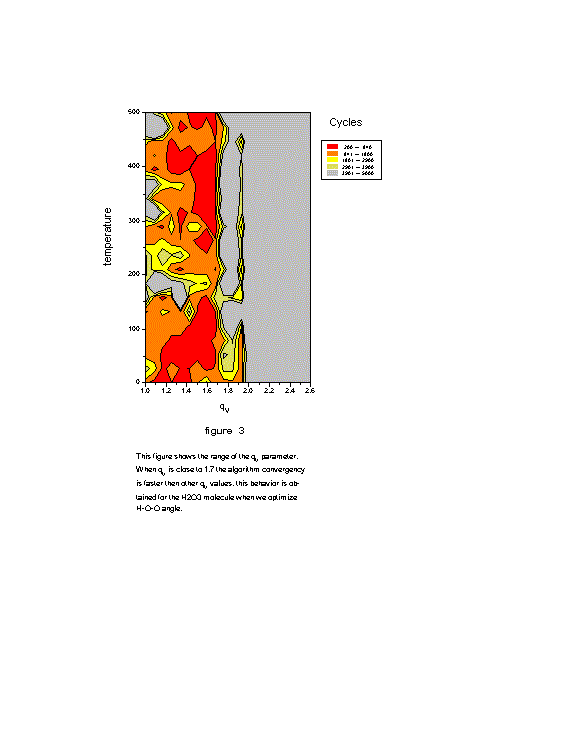
The figure 3 shows the range of the q ![]() parameter. When q
parameter. When q ![]() is close to 1.6 the algorithm convergence is faster than other q
is close to 1.6 the algorithm convergence is faster than other q ![]() values. This behavior is obtained for the H2O3
molecule when we optimize H-O-O angle.
values. This behavior is obtained for the H2O3
molecule when we optimize H-O-O angle.
Both results, in case of equilibrium geometry, according
qualitatively which a semi-empirical procedure [5],
however we have obtained all mapping using SMO faster than semi-empirical
one.
Other important result is that the variation range of q (Fig. 2-3) agree with the relationship proposed in reference [14], that is;
That equation defines the region where q ![]() is optimized, wich corresponds to a minimum number of steps to find the
global minimum. D is the space dimension or the number of parameters
to be optimized.
is optimized, wich corresponds to a minimum number of steps to find the
global minimum. D is the space dimension or the number of parameters
to be optimized.
POLYPEPTIDES (Dimension D=36)
As a second application we have used the SMO approach
to verify the formation of ![]() -helix structure in polypeptides. Other goal of this application is to
check the potentiality of the SMO method in case of large molecular system
or in case o large dimension D. We investigate two structures: pentalanin
and icoalanin molecules. These are polypeptides molecules, composed by
five and twenty alanin residues, respectively.
-helix structure in polypeptides. Other goal of this application is to
check the potentiality of the SMO method in case of large molecular system
or in case o large dimension D. We investigate two structures: pentalanin
and icoalanin molecules. These are polypeptides molecules, composed by
five and twenty alanin residues, respectively.
The search of the global minimum by the SMO approach allow
us to map the hypersurface's conformational energy, as shown in Figures
4 and 5 for the H ![]() O
O ![]() and icoalanin molecules, respectively.
and icoalanin molecules, respectively.
The ![]() helix (a polypeptide structure) is a rodlike structure. The tightly coiled
polypeptide main chain forms the inner part of the rod, and the side chain
extends outward in a helical array in low dielectric medium. The
helix (a polypeptide structure) is a rodlike structure. The tightly coiled
polypeptide main chain forms the inner part of the rod, and the side chain
extends outward in a helical array in low dielectric medium. The ![]() helix is stabilized by hydrogen bonds between the NH and CO groups of the
main chain. The CO group of each amino acid is hydrogen bonded to the NH
group of the amino acid that is situated four residues ahead in the linear
sequence. Thus, all the main-chain CO and NH groups are hydrogen bonded.
We have studied these compounds, optimizing the dihedral angles:
helix is stabilized by hydrogen bonds between the NH and CO groups of the
main chain. The CO group of each amino acid is hydrogen bonded to the NH
group of the amino acid that is situated four residues ahead in the linear
sequence. Thus, all the main-chain CO and NH groups are hydrogen bonded.
We have studied these compounds, optimizing the dihedral angles: ![]() (CH
(CH ![]() C-N-CH
C-N-CH ![]() ),
), ![]() (N-CH
(N-CH ![]() C-N) and
C-N) and ![]() (C- N-CH
(C- N-CH ![]() C).
C).
We show that pentalanin does not form stable ![]() helix structure, probably because the number of hydrogen bonds are insufficient
to maintain the helical array. We optimized the
helix structure, probably because the number of hydrogen bonds are insufficient
to maintain the helical array. We optimized the ![]() ,
, ![]() and
and ![]() dihedral angles and obtained, using SMO approach, a global minimum which
does not correspond to the helical array,.also the ''most frequent''
dihedral angles and obtained, using SMO approach, a global minimum which
does not correspond to the helical array,.also the ''most frequent'' ![]() ,
, ![]() and
and ![]() dihedral angles found in the THOR-GSA run does not correspond to
dihedral angles found in the THOR-GSA run does not correspond to ![]() helix structures.
helix structures.
On the contrary, the icoalanin molecule is an ![]() helix structure. The following improper angles are expected for an
helix structure. The following improper angles are expected for an ![]() helix structure:
helix structure: ![]() = 180
= 180 ![]() ,
, ![]() = - 40
= - 40 ![]() and
and ![]() = - 60
= - 60 ![]() . The
. The ![]() torsion angle has two minima. One at 0
torsion angle has two minima. One at 0 ![]() and the other one at 180
and the other one at 180 ![]() . The later value is always observed in biological structures, except for
special situations for proline residues. In order to simplify the calculation
we fix the
. The later value is always observed in biological structures, except for
special situations for proline residues. In order to simplify the calculation
we fix the ![]() = 180
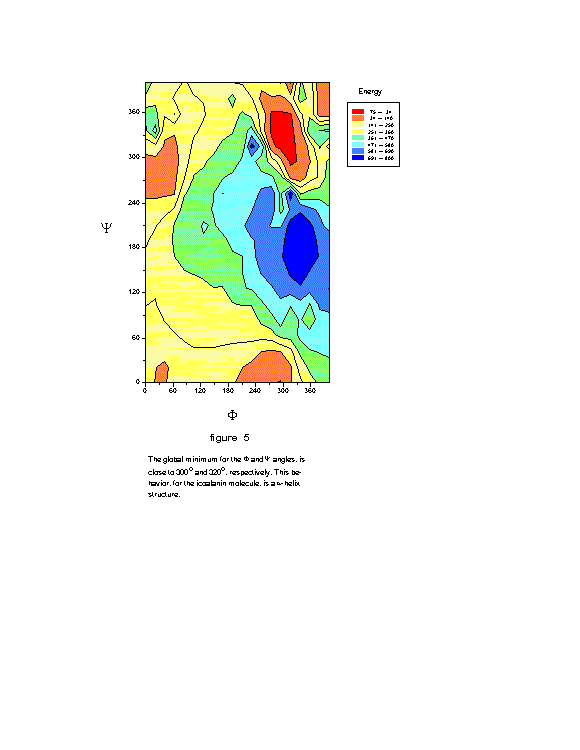
= 180 ![]() . Figure 5 shows the
. Figure 5 shows the ![]() ,
, ![]() conformation energy surface obtained. This figure shows, as expected, that
the global minimum occurs at the angles
conformation energy surface obtained. This figure shows, as expected, that
the global minimum occurs at the angles ![]() = -60
= -60 ![]() and
and ![]() = -40
= -40 ![]() .
.
We can obtain the ![]() helix structure, simulating all
helix structure, simulating all ![]() ,
, ![]() pairs, in a few minutes using an IBM risc-6000 workstation. With q
pairs, in a few minutes using an IBM risc-6000 workstation. With q ![]() =1.5, q
=1.5, q ![]() = 2.0 and T=100 the simulation takes 3673 to 100000 cycles for 18
= 2.0 and T=100 the simulation takes 3673 to 100000 cycles for 18 ![]() ,
, ![]() pairs (D=36). Meanwhile, using MD, we would have waited on a few days of
simulation!! Further, depending on the initial condition, we do not obtaining
an
pairs (D=36). Meanwhile, using MD, we would have waited on a few days of
simulation!! Further, depending on the initial condition, we do not obtaining
an ![]() helix structure since MD only guarantees a dynamical structure which is
frequently a local minimum, because protein folding is not in MD time scale.
helix structure since MD only guarantees a dynamical structure which is
frequently a local minimum, because protein folding is not in MD time scale.
We would like to point out, in order to compare the SMO
method with the usual Monte Carlo one, that a similar simulation was done
in reference [16]. In this last case the folding
of ![]() helix is reached in 100 millions of cycles, which makes the SMO
a promising method for protein folding studies.
helix is reached in 100 millions of cycles, which makes the SMO
a promising method for protein folding studies.
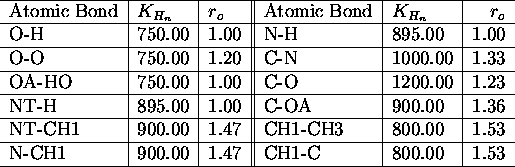
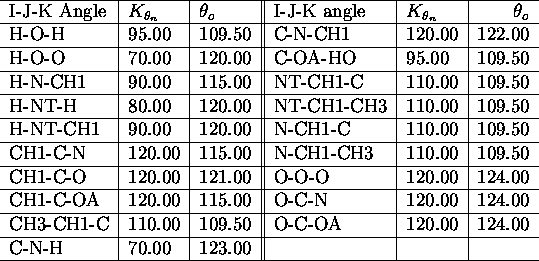
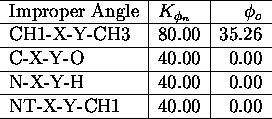
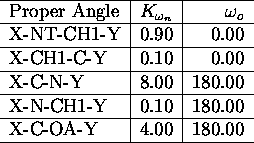
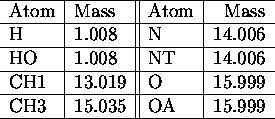
All force field parameters used in the applications presented
in this paper, are listed in the tables I. a.,b
,c,d,e,
The used atomic nomenclature is H (hydrogen), HO (hydroxyl hydrogen), CH
![]() (aliphatic CH-group), CH
(aliphatic CH-group), CH ![]() (aliphatic CH
(aliphatic CH ![]() -group), N (nitrogen), NT (terminal nitrogen), O (oxygen), OA (hydroxyl
oxygen). \
-group), N (nitrogen), NT (terminal nitrogen), O (oxygen), OA (hydroxyl
oxygen). \
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}