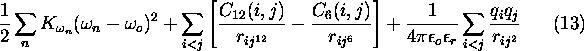Next: APPLICATIONS Up: STOCHASTIC MOLECULAR OPTIMIZATION USING Previous: GENERALIZED SIMULATED ANNEALING
![]()
![]()
![]()
Next: APPLICATIONS
Up: STOCHASTIC
MOLECULAR OPTIMIZATION USING Previous: GENERALIZED
SIMULATED ANNEALING
The THOR program was developed to be a comprehensive and flexible tool to investigate macromolecular structure of biological interest like proteins and membranes. The computational code is based in a classical force field. Both Molecular Dynamics and Optimization gradient methods are available. Now THOR is also able to attain conformational structures with SMO methods. The choice of either of these methods is given by the user's necessity. If we need dynamics properties (a local minimum structure) we can use MD (gradient approach). If it is better to use the conformation energy hypersurface mapping or the global minimum localization, we must choose the SMO method.
It is well know that the biological activity depends on the spatial conformation acquired by macromolecules in the physiological medium. The action of hormones and drugs is also dependent on molecular structure of the target molecules. In recent years, models developed for biological molecules allowed [13] significant advances by proposing new molecular architectures able to satisfy specific properties, increasing their biological efficiency.
Most of molecular modeling has been restricted to molecules independent of environmental solvent. However many important biological functions are related to the nature and to the heterogeneity of the solvent medium. For example, proteins which compose membranes depend at the molecular level on the inhomogeneous medium composed by the membrane and its neighborhood. In biological mem- branes, fundamental molecular properties are associated to the conformational changes brought about by changes in the solvent medium, i.e., between the membrane lipidic phase and its adjacent medium water phase. Therefore, THOR is able to sim- ulate this interface and to insert molecules into this heterogeneous medium [3]. Further, the THOR is able to accomplish this for isolate molecules as well as for complex sets of molecules, for example, simulating a membrane structure or enzyme-inhibitor complexes.
The THOR molecular interaction is simulated by a classical field, parameterized
with quantum and/or empirical data. The con- formational energy of the
molecule is made up of a sum of bond and nonbond terms. In this approach,
only the covalently hydrogen bonded to oxygen or to nitrogen are considered
explicitly, while the CH ![]() , CH
, CH ![]() and CH
and CH ![]() groups are assumed to be an atomic unit.
groups are assumed to be an atomic unit.
The THOR force field is:
![]()
![]()
where ![]() is the bond energy term,
is the bond energy term, ![]() is the angular energy term,
is the angular energy term, ![]() is the proper dihedral energy term,
is the proper dihedral energy term, ![]() is the improper dihedral energy term,
is the improper dihedral energy term, ![]() is the Van der Waals energy term,
is the Van der Waals energy term, ![]() is the Coulomb energy term.
is the Coulomb energy term.
In the following section we present applications for the SMO approach.