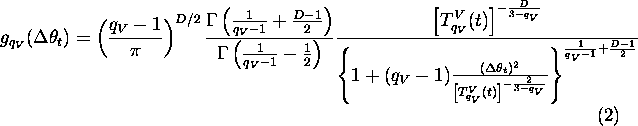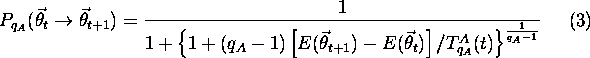Next: Applications Up: Geometry Optimization and Conformational Previous: Introduction
![]()
![]()
![]()
Next: Applications
Up: Geometry
Optimization and Conformational Previous: Introduction
Here, we implement the GSA algorithm on a semi-empirical quantum method to calculate the minimal energy conformational geometry for different molecular structures.This technique can be indifferently applied on all ''ab-initio '' or semi-empirical quantum methods. We have used, in the present case, a semi-empirical one only for computational convenience.
The GSA method is based on the correlation between the minimization of a cost function (molecular energy) and the geometries randomly obtained through a slow cooling. In this technique, an artificial temperature is introduced and gradually cooled, in complete analogy with the well known annealing technique frequently used in metallurgy when a molten metal reaches its crystalline state (global minimum of the thermodynamical energy). In our case the temperature is intended as an external noise.
The procedure consists in comparing the total semi-empirical energies for two random geometries obtained from the GSA routine. The artificial temperature ( or set of temperatures) acts as a source of stochasticity extremely convenient for eventually detrapping from local minima. Near the end of the process, the system hopefully is inside of the attractive basin of the global minimum ( or in one of global minima, if there is degeneracy). The challenge is to get cool the temperature the quickest we can but still having the guarantee that no irreversible trapping at any local minimum occurs. More precisely speaking, we search for the quickest annealing (that is, in some sense approaching a quenching) which preserves the probability of ending in a global minimum being equal one.
The present GSA routine was built using the same procedure presented
in [8]. We apply this algorithm in order to
study a set of molecules which present one or more different conformations
by rotating dihedral angles ( ![]() ) around the X-Y bonds. In summary the whole algorithm for mapping the
global minimum of the energy function is:
) around the X-Y bonds. In summary the whole algorithm for mapping the
global minimum of the energy function is:
\
(i) Fix the parameters ![]() (we recall that
(we recall that ![]() and (1,2) respectively correspond to the Boltzmann and Cauchy machines).
Start, at t=1, with an arbitrary value
and (1,2) respectively correspond to the Boltzmann and Cauchy machines).
Start, at t=1, with an arbitrary value ![]() and a high enough value
and a high enough value ![]() ( visiting temperature) and cool as follows:
( visiting temperature) and cool as follows:
where t is the discrete time corresponding to the computer iteration,
and ![]() (
( ![]() ) is the acceptance index (visiting index).
) is the acceptance index (visiting index).
\
(ii) Then randomly generate ![]() from
from ![]() by using the visiting distribution probability
by using the visiting distribution probability ![]() as
as
with -180< ![]()
![]() ;
; ![]() is the gamma function; D is the number of components of
is the gamma function; D is the number of components of ![]() . This procedure assures that the system can both escape from any local
minimum and explore the entire energy surface.
. This procedure assures that the system can both escape from any local
minimum and explore the entire energy surface.
\
(iii) Then calculate the total electronic energy ![]() by using the MOPAC program:
by using the MOPAC program:
If ![]()
![]() , replace
, replace ![]() by
by ![]()
If ![]() , run a random number
, run a random number ![]() if
if ![]() (acceptance probability) given by
(acceptance probability) given by
with ( ![]() ), retain
), retain ![]() ; otherwise, replace
; otherwise, replace ![]() by
by ![]() .
.
\
(iv) Calculate the new temperature ![]() using Eq.(1) and go back to (ii) until the minimum of
using Eq.(1) and go back to (ii) until the minimum of ![]() is reached within the desired precision.
is reached within the desired precision.
\
In short, this computational method is based on a stochastic dynamics
which enables, with probability one, the identification of a global minimum
of the energy hyper-surface, which depends on a continuous D-dimensional
variable ![]() , (in this paper
, (in this paper ![]() are dihedral angles ). While the number t of computational iterations
increases, it might happen that
are dihedral angles ). While the number t of computational iterations
increases, it might happen that ![]() provisorily stabilizes on a given value, and eventually abandons it running
towards the global minimum. This temporary residence can be used, as a
bonus of the present method, to identify some of the local minima. The
ordinate (Number of cycles), in the figures 1 to 4, represents the
frequency (temporary residence) of the positive trials when a tested angle
appears.
provisorily stabilizes on a given value, and eventually abandons it running
towards the global minimum. This temporary residence can be used, as a
bonus of the present method, to identify some of the local minima. The
ordinate (Number of cycles), in the figures 1 to 4, represents the
frequency (temporary residence) of the positive trials when a tested angle
appears.
In figures 1 to 4 ( D=1 case) we observe the arising of some dihedral angles (noises) which do not represent the searched local or global minima. They appear with minor frequency, and in order to eliminate this noises it is convenient to repeat the procedure (i) to (iv) using different initial conditions. In this case we can also verify that all degenerate minima will be visited with the same frequency.
In the figure 6 we present the result obtained for the molecule H ![]() O
O ![]() (Fig.5) with two parameters to be optimized (D=2), i.e.,
(Fig.5) with two parameters to be optimized (D=2), i.e., ![]() .
.