| THOR+DVM Procedure |
It is based on a hybrid approach which makes use of Classical Molecular Dynamics (MD) and Stochastic Molecular Dynamics Monte Carlo (MC) simulations, coupled to Embedded Cluster Density Functional (ECDF) algorithms.
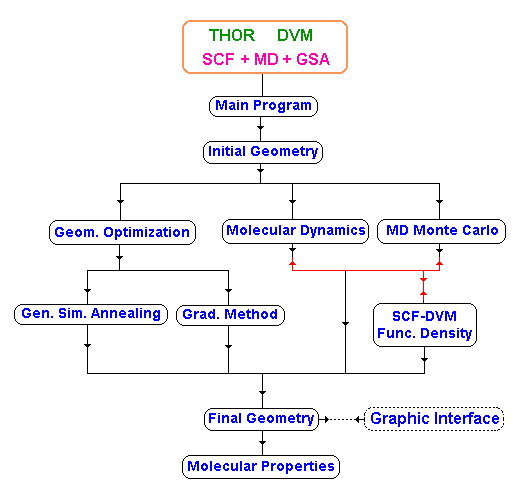
In this scheme, parameterized interatomic potentials are used in MD/MC to relax the defect or interface structure, subject to constrains imposed by electron microscopy and other experimental data. See flowchart in Fig.1.
The methodology used in our molecular dynamic approach is to run the MD program simultaneously with the DVM code. During the dynamic process the MD routine call the SCF-DVM program to redefine the atomic charge. In case of ionic crystals this kind of procedure is necessary to guarantee the time dependence of the atomic charge. In general, the results produced by the DVM code are used to adjust the classical force field. See flowchart in Fig.1.
In parallel with this project we are developing a Stochastic Molecular Mechanics procedure, based on Monte Carlo method, to study some thermodynamic properties in crystals. In this case, random molecular geometries are calculated and used by the Simulated Annealing Method (GSA) to map the molecular energy hyper-surface, as well to study the atomic diffusion.
The simulation package comes with the THOR force field and SCF-DVM has the purposes:
- Energy minimization of arbitrary
molecules (solids, crystals, proteins, polymers, nucleotides, sugars, etc.)
- Analysis of conformations obtained
by experiment, theoretical quantum method or by computer simulation
- Simulations at biological interface
(membrane/water) with dielectric discontinuity
- Diffusion of impurities in solids
- Defects and interface structure
in solids
- First-principle calculations
are carried out at equilibrium, metastable, near-equilibrium geometries,
and along transition path.
- Diffusion of solute in molecular
sieves (Zeolites)
- Prediction of the dependence
of a molecular conformation on the type of environment (water, crystal,
etc.)
- Mechanical stress in the crystal,
- Thermodynamic properties,
- Grain-boundary,
- Evaporation in crystal
{kind=link}